Содержание
| Мальформация Арнольда — Киари | |
|---|---|
 |
|
| МКБ-10 | Q 07.0 07.0 |
| МКБ-10-КМ | Q07.0 |
| МКБ-9 | 741.0 741.0 |
| OMIM | 207950 |
| DiseasesDB | 899 |
| MeSH | D001139 |
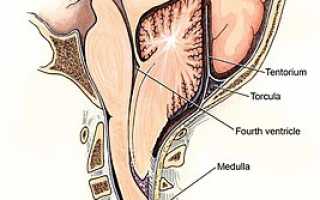
Мальформация Арнольда — Киари — это состояние, при котором миндалины мозжечка опускаются в большое затылочное отверстие, что приводит к сдавливанию продолговатого мозга. В некоторых случаях (мальформация Киари 2) наблюдаются также гидроцефалия, сирингомиелия и менингомиелоцеле. Симптомы этого заболевания могут варьироваться в зависимости от степени поражения продолговатого мозга и мозжечка, включая затылочные боли, нарушение глотания и атаксию. Оно также может сочетаться с базилярной импрессией или инвагинацией, ассимиляцией атланта.
В норме миндалины мозжечка находятся выше большого затылочного отверстия. У пациентов с аномалией Арнольда-Киари миндалины мозжечка смещаются вниз до уровня первого или даже второго шейного позвонка, что препятствует нормальному току спинномозговой жидкости.
Ранее считалось, что аномалия Арнольда-Киари всегда является врожденной, однако сейчас исследователи полагают, что у большинства людей смещение миндалин мозжечка происходит во время активного роста мозга, когда кости черепа медленно развиваются. Только небольшое количество пациентов с аномалией Арнольда-Киари действительно имеют врожденную форму этого заболевания. Кроме того, существуют и другие врожденные заболевания, которые могут привести к смещению миндалин мозжечка, такие как платибазия, базилярная инвагинация, аномалия Денди-Уокера и другие.
С 2005 года существует новая теория, согласно которой причиной Мальформации или Синдрома Арнольда Киари 1 является аномальное натяжение спинного мозга из-за напряженной концевой нити. Эта теория также связывает натяжение спинного мозга с другими проблемами, которые часто возникают вместе с Синдромом Арнольда Киари 1, такими как идиопатическая сирингомиелия, идиопатический сколиоз, платибазия, базиллярная импрессия и другие.
Частота встречаемости этого заболевания составляет от 3.3 до 8.2 случаев на 100000 человек.
Сирингомиелия развивается у 80% пациентов, страдающих от этого заболевания.
Средний возраст пациентов составляет 25-40 лет.
Типы [ править | править код ]
Типы Синдрома Арнольда Киари (САК) [1] :
САК.I. Миндалины мозжечка опущены без каких-либо других мальформаций нервной системы.
САК.II. Миндалины мозжечка опущены с нейропозвоночной мальформацией, при которой спинной мозг прикреплен к позвоночному каналу.
 Читайте также:
Читайте также:

САК.III. Миндалины мозжечка опущены с затылочной энцефалоцеле и аномалиями мозга при САК.II.
САК.IV. Миндалины мозжечка отсутствуют или имеют гипоплазию, связанную с отсутствием намёта мозжечка.
САК.0. В настоящее время зарегистрированы случаи клинической картины, характерной для САК.I, без опущения миндалин мозжечка.
САК.1.5. Недавно описан САК.1,5 с опущением миндалин мозжечка и ствола головного мозга в затылочное отверстие.
На основе анализа клинико-рентгенологических и нейровизуализационных наблюдений выделены 3 варианта САК I: передний, промежуточный и задний (С. В. Можаев и соавт., Институт мозга человека РАН, Санкт-Петербург, кафедра неврологии и нейрохирургии СПбГМУ, 2007) [2] :
- передний вариант включает отклонение зуба С2 позвонка назад, платибазию или базилярную импрессию, а также нависание продолговатого мозга над зубовидным отростком;
- промежуточный вариант предполагает элементы компрессии вентральных отделов продолговатого и верхнешейных сегментов спинного мозга зубовидным отростком С2 позвонка и дорсальных — сместившимися миндалинами мозжечка;
- задний вариант предполагает элементы компрессии дорсальных отделов продолговатого и верхнешейных сегментов спинного мозга смещенными в большое затылочное отверстие миндалинами мозжечка.
Лечение [ править | править код ]
Подзатылочная декомпрессионная краниотомия (ПДК) не устраняет источник заболевания. ПДК при Синдроме Арнольда Киари I (САК.I) просто снимает давление с нервной системы в затылочном отверстии, что иногда может привести к временному улучшению состояния пациента после операции.
Этот метод лечения сопровождается высоким риском осложнений и смертности (0,7-12 %), что может быть оправдано в случаях, когда смертность от заболевания сама по себе выше, например, при опухолях, сосудистых аномалиях или гематомах затылочного отверстия. Однако риск внезапной смерти от САК.I значительно ниже, чем риск смерти от предлагаемого лечения. За последние три десятилетия было описано всего 8 случаев внезапной смерти от заболевания:
«… в литературе показатели смертности варьируют от 0,7 % (АГАХАНИ, Дж.Н. и др., 1999) до 1,4 % (ПОЛ, К.С. и др., 1983) и 12,1 % (ЛОРЕНЦО, Д.Н. и др., 1982). Два пациента из нашей серии умерли в раннем послеоперационном периоде, что свидетельствует о хирургической смертности 1 %…» (КЛЕКАМП, Дж.; САМИ, М., 2012).
Рассечение концевой нити при опущении миндалин мозжечка или САК.I гораздо менее травматично, чем традиционное лечение (открытие задней черепной ямки при ПДК). Использование минимально инвазивной техники при рассечении концевой нити не сопровождается никакими осложнениями и смертностью.
-
Читайте также:

Военкомат [ править | править код ]
В расписании заболеваний, освобождающих от военной службы (Постановление Правительства РФ от 04.07.2013 N 565 (ред. от 21.04.2018)), не указана мальформация Арнольда-Киари.
Педиатр Анна Колинько рассказывает о патологии развития головного мозга, которая может встречаться у 30% населения.
Синдром хронической усталости, головокружения и боли в шее могут быть результатом мальформации (аномалии) Арнольда-Киари. С появлением широкого использования МРТ стало ясно, что это заболевание встречается у 14-30% населения.
Мальформация Арнольда-Киари (МАК) – это патология развития ромбовидного мозга, включающая продолговатый и задний мозг, а также Варолиев мост и мозжечок. При МАК задняя черепная ямка не соответствует мозговым структурам, расположенным в этой области: мозжечок и продолговатый мозг опускаются ниже большого затылочного отверстия из-за их небольших размеров, что приводит к их сдавливанию и нарушению циркуляции жидкости внутри черепа. МАК относится к группе кранио-вертебральных (черепно-позвоночных) мальформаций.
В эпоху до МРТ частота МАК оценивалась от 3,3 до 8,2 случаев на 100 000 населения, а у новорожденных – 1 случай на 4-6 тысяч. Сегодня стало ясно, что распространенность синдрома Арнольда-Киари гораздо выше. Из-за бессимптомного течения и учета разных типов МАК, цифры очень разнятся – от 14 до 30%.
Все первые описания мальформации были после смерти. В 1883 году шотландский анатом Джон Клеланд (J. Cleland, 1835-1925) впервые описал удлинение ствола и опущение миндалин мозжечка в большое затылочное отверстие у 9 умерших новорожденных. В 1891 году австрийский патолог Ганс фон Киари (H. Chiari, 1851-1916) подробно охарактеризовал 3 типа мальформации у детей и взрослых. А в 1894 году немецкий патолог Юлиус Арнольд (J. Аrnold, 1835-1915) подробно описал синдром Киари 2 типа, в сочетании со спинномозговой грыжей (spina bifida). В 1896 году Киари дополнил свою классификацию четвертым типом. В 1907 году ученики Арнольда использовали термин "мальформация Арнольда-Киари" для обозначения аномалии 2 типа. Теперь это название распространилось на все типы. Некоторые врачи справедливо отмечают, что вклад Арнольда немного преувеличен и правильным будет термин "мальформация Киари".
Версии о причинах
Этиология и патогенез синдрома Арнольда — Киари до сих пор остаются неясными. Киари предположил, что смещение мозжечка и продолговатого мозга происходит из-за внутриэмбриональной гидроцефалии, которая возникает в результате стеноза сильвиева водопровода — узкого канала длиной 2 см, соединяющего III и IV желудочки мозга.
Клеланд считал, что аномалия связана с первичным недоразвитием ствола головного мозга. В 1938 году канадский нейрохирург Уайлдер Пенфилд (W. G. Penfield, 1891–1976 гг.) и его коллега предложили "теорию тяги": в процессе роста фиксированный спинной мозг втягивает в полость позвоночного канала расположенные выше отделы. В "унифицированной" теории Дэвид Маклон (D. G. McLone) и Пол Неппер (P. A. Knepper) в 1989 году предположили, что первично возникает дефект нервной трубки с истечением ликвора и недостаточным расширением желудочковой системы, что приводит к формированию уменьшенной задней черепной ямки. Однако последующие исследования указывают на то, что существуют различные варианты патологии Арнольда — Киари: с уменьшением задней черепной ямки и без такового, с нарушением ликворооттока и без него. Описаны случаи МАК 2 типа в семьях, однако роль генетических факторов до сих пор не полностью изучена.
Типы мальформаций
1 тип — перенос миндалин мозжечка в позвоночный канал ниже уровня большого затылочного отверстия без наличия спинномозговой грыжи. У 15–20 % пациентов этот тип сочетается с гидроцефалией, а у 50 % больных — с сирингомиелией — заболеванием, при котором в спинном и продолговатом мозге образуются полости. В 1991 году было предложено разделить аномалии Арнольда — Киари 1 типа на тип А — с сирингомиелией и тип В — без сирингомиелии.
-
Читайте также:

Сирингомиелии при Арнольде — Киари 1 степени.
Энцефаломенингоцеле — врожденная грыжа головного мозга и его оболочек, содержащая цереброспинальную жидкость.
Спинальная дизрафия — порок развития, заключающийся в отсутствии слияния по средней линии парных закладок кожи, мускулатуры, позвонков, спинного мозга
2 тип — перенос нижних отделов червя мозжечка, продолговатого мозга и IV желудочка. Отличительным признаком данного типа является сочетание со спинномозговой грыжей (spina bifida) в поясничном отделе, отмечается прогрессирующая гидроцефалия, часто — стеноз водопровода мозга. Среди детей с менингомиелоцеле до 90 % случаев сопровождается аномалией Арнольда — Киари 2 степени.
0, 1 и 2 степени синдрома Арнольда — Киари наиболее распространены в популяции. III и IV типы обычно несовместимы с жизнью.
Симптоматика
Неврологические проявления аномалии Арнольда-Киари 0 и 1 типов обычно возникают в возрасте от 20 до 40 лет. Смещение миндалин мозжечка может усиливаться под воздействием неблагоприятных факторов. Чаще всего пациенты с аномалией Арнольда-Киари 0 типа жалуются на головную боль, особенно в области шеи, а также на боль в шее. У взрослых с аномалией Арнольда-Киари 1 типа часто наблюдаются нистагм, дизартрия, атаксия, интенционный тремор (тремор при произвольных движениях), головная боль, головокружение, нарушение чувствительности, парезы, нарушение функции тазовых органов, нарушения частоты и ритма пульса, ритма дыхания, лабильность артериального давления, а также симптомы поражения каудальной группы черепных нервов (IX, X, XI, XII пары) – нарушение чувствительности лица и бульбарные расстройства (расстройства глотания и речи).
Синдром Арнольда-Киари 2 степени впервые проявляется не у взрослых, а у новорожденных или в раннем детском возрасте. Аномалия Арнольда-Киари 2 типа характеризуется более тяжелым течением, и дети с таким диагнозом уже рождаются с гидроцефальной формой черепа. Гидроцефалия мешает нормальному развитию. Кроме того, у таких детей наблюдаются нарушения дыхания, сердцебиения и глотания. Часто заболевание сопровождается судорожными припадками. У детей развивается нистагм, апноэ, стридор, парез голосовых связок, дисфагия с регургитацией, нарушение тонуса в конечностях. Симптомы неврологического характера в первую очередь зависят от нарушений ликвородинамики, а не от степени смещения миндалин мозжечка.
Терапия
Лечение аномалий Арнольда-Киари зависит от степени проявления неврологических симптомов. Консервативная терапия включает применение препаратов, которые снижают воспаление, а также миорелаксантов. Если консервативное лечение не дает результатов в течение 2-3 месяцев или у пациента имеется серьезный неврологический дефицит, может потребоваться операция. Во время операции устраняется сдавление нервных структур и восстанавливается нормальный поток жидкости путем расширения задней черепной ямки и установки шунта. Оперативное лечение эффективно в 50-85% случаев, в остальных случаях симптомы могут не полностью исчезнуть. Рекомендуется проводить операцию до развития тяжелого неврологического дефицита, так как восстановление происходит лучше при минимальных изменениях неврологического состояния. Такие операции проводятся во многих нейрохирургических центрах России и входят в программу высокотехнологичной медицинской помощи по системе ОМС.
Пациенты с мальформацией Арнольда-Киари 0 и 1 типа могут даже не знать о наличии этого заболевания в течение всей жизни. Благодаря пренатальной диагностике МАК II, III и IV типа, дети с этой патологией рождаются все реже, а современные методы лечения позволяют им жить дольше.
Аномалия Арнольда-Киари – это развитие, при котором размеры черепной ямки и структурных элементов мозга несоответствуют друг другу. Мозжечковые миндалины опускаются ниже обычного уровня и могут быть сдавлены.
-
Читайте также:

Симптомы аномалии Арнольда-Киари проявляются в виде частых головокружений и иногда могут привести к инсульту. Признаки аномалии могут быть отсутствовать долгое время, а затем внезапно появиться, например, после вирусной инфекции, удара головы или других факторов. Это может произойти в любом возрасте.
Описание болезни
Сущность данного заболевания заключается в неправильной позиции продолговатого мозга и мозжечка, что приводит к появлению краниоспинальных синдромов. Врачи часто ошибочно диагностируют его как атипичный случай сирингомиелии, рассеянного склероза или спинномозговой опухоли. У большинства пациентов с аномалией развития ромбэнцефалона также наблюдаются другие нарушения в спинном мозге, такие как кисты, которые вызывают быстрое разрушение спинномозговых структур.
Название этого заболевания происходит от имен патологоанатома Арнольда Джулиуса (Германия), который описал аномальное отклонение в конце 18 века, и врача из Австрии Ганса Киари, который изучал это заболевание в то же время. Распространенность этого нарушения варьирует от 3 до 8 случаев на каждые 100000 человек. В основном встречаются аномалии Арнольда Киари 1 и 2 степени, а взрослые с 3-м и 4-м типами этой аномалии обычно живут недолго.
Аномалия Арнольда Киари 1 типа проявляется в опущении элементов задней черепной ямки в спинальный канал. Болезнь Киари 2 типа характеризуется изменением положения продолговатого мозга и четвертого желудочка, и часто сопровождается водянкой. Третья степень патологии встречается гораздо реже и характеризуется выраженными смещениями всех элементов черепной ямки. Четвертый тип представляет собой дисплазию мозжечка без его смещения вниз.
Причины заболевания
По данным некоторых авторов, болезнь Киари представляет собой недоразвитие мозжечка, которое сопровождается различными отклонениями в других частях мозга. Самая распространенная форма этого нарушения – аномалия Арнольда Киари 1 степени. Она характеризуется опусканием миндалин мозжечка в спинальный канал, что может произойти из-за перемещения продолговатого мозга вниз. Часто эта патология сопровождается нарушениями краниовертебральной границы.
Клинические проявления болезни могут появиться только в возрасте 30-40 лет. Важно отметить, что бессимптомное наличие опущенных миндалин мозжечка не требует лечения и часто обнаруживается случайно при проведении МРТ. На сегодняшний день причины и механизм развития этой болезни плохо изучены. Генетический фактор играет определенную роль в ее возникновении.
Механизм развития болезни можно разделить на три составляющих:
- генетически обусловленная врожденная остеоневропатия;
- травматизация ската во время родов;
- повышенное давление ликвора на стенки спинномозгового канала.
Проявления
По частоте появления выделяют следующие признаки:
- головные боли – у трети пациентов;
- боль в конечностях – 11%;
- слабость в руках и ногах (в одной или двух конечностях) – больше половины пациентов;
- чувство онемения в конечности – половина больных;
- снижение или потеря температурной и болевой чувствительности – 40%;
- шаткость походки – 40%;
- непроизвольные колебания глаз – треть больных;
- двоение в глазах – 13%;
- нарушения глотания – 8%;
- рвота – у 5%;
- нарушения произношения – 4%;
- головокружения, глухота, онемение в лицевой области – у 3% больных;
- синкопальные (обморочные) состояния – 2%.
Болезнь Киари второй степени (диагностируется у детей) объединяет дислокацию мозжечка, ствола и четвертого желудочка. Неотъемлемым признаком является наличие менингомиелоцеле в области поясницы (грыжа спинального канала с выпячиванием вещества спинного мозга). Неврологическая симптоматика развивается на фоне аномального строения затылочной кости и шейного отдела позвоночного столба. Во всех случаях присутствует гидроцефалия, часто – сужение водопровода мозга. Неврологические признаки появляются с самого рождения.
Операция при менингомиелоцеле проводится в первые дни после рождения. Последующее хирургическое расширение задней черепной ямки позволяет достичь хороших результатов. Многие пациенты нуждаются в шунтировании, особенно при стенозе Сильвиевого водопровода. При аномалии третьей степени черепно-мозговая грыжа внизу затылка или в верхней шейной области сочетается с нарушениями развития мозгового ствола, краниального основания и верхних позвонков шеи. Образование захватывает мозжечок и в 50% случаев – затылочную долю.
Эта патология встречается очень редко, имеет неблагоприятный прогноз и резко сокращает продолжительность жизни даже после операции. Точное количество времени, которое человек будет жить после своевременного вмешательства, невозможно сказать, но, вероятнее всего, недолго, так как эта патология считается несовместимой с жизнью. Четвертая степень заболевания представляет собой отдельную гипоплазию мозжечка и на сегодняшний день не относится к симптомокомплексам Арнольда-Киари.
Клинические проявления при первом типе прогрессируют медленно, в течение нескольких лет и сопровождаются включением в процесс верхнего шейного спинномозгового отдела и дистального отдела продолговатого мозга с нарушением работы мозжечка и каудальной группы черепных нервов. Таким образом, у лиц с аномалией Арнольда-Киари выделяют три неврологических синдрома:
- Бульбарный синдром сопровождается нарушением функции тройничного, лицевого, преддверно-улиткового, подъязычного и вагусного нервов. При этом наблюдаются нарушения глотания и речи, нистагм, головокружения, расстройства дыхания, парез мягкого неба с одной стороны, охриплость голоса, атаксия, дискоординация движений, неполный паралич нижних конечностей.
- Сирингомиелитический синдром проявляется атрофией мышц языка, нарушением глотания, отсутствием чувствительности в лицевой области, хриплостью голоса, нистагмом, слабостью в руках и ногах, повышением мышечного тонуса и т. д.
- Пирамидный синдром характеризуется незначительным спастическим парезом всех конечностей с гипотонусом рук и ног. Сухожильные рефлексы на конечностях повышаются, брюшные рефлексы не вызываются или снижаются.
Боли в области затылка и шеи могут усиливаться при кашле, чихании. В руках снижается температурная и болевая чувствительность, а также мышечная сила. Часто возникают обмороки, головокружения, ухудшение зрения у больных. При запущенной форме появляются апноэ (кратковременная остановка дыхания), быстрые неконтролируемые движения глаз, ухудшение глотательного рефлекса.
Интересным клиническим признаком у таких людей является вызывание симптомов (синкопии, покалывания, боли и т. д.) напряжением, смехом, кашлем, пробой Вальсальвы (усиленное выдохивание при закрытом носу и рте). При усилении очаговых симптомов (стволовых, мозжечковых, спинномозговых) и гидроцефалии возникает вопрос о хирургическом расширении задней черепной ямки (субокципитальная декомпрессия).
Диагностика
Диагноз аномалии первого типа не сопровождается повреждением спинного мозга и устанавливается в основном у взрослых с помощью КТ и МРТ. Согласно результатам патологоанатомического вскрытия, у детей с грыжей спинномозгового канала болезнь Киари второго типа обнаруживается в большинстве случаев (96–100%). С использованием УЗИ можно выявить нарушения циркуляции ликвора. В норме цереброспинальная жидкость свободно циркулирует в подпаутинном пространстве.
Боковой рентген и МР картина черепа показывает расширение канала позвоночного столба на уровне С1 и С2. На ангиографии сонных артерий наблюдается огибание миндалины мозжечковой артерией. На рентгене отмечаются такие сопутствующие изменения краниовертебральной области, как недоразвитие атланта, зубовидного отростка эпистрофея, укорачивание атлантозатылочной дистанции.
При сирингомиелии на боковом снимке рентгена наблюдается недоразвитие задней дуги атланта, недоразвитие второго шейного позвонка, деформация большого затылочного отверстия, гипоплазия боковых частей атланта, расширение позвоночного канала на уровне С1-С2. Дополнительно следует провести МРТ и инвазивное рентгенологическое исследование.
Манифестация симптомов болезни у взрослых и лиц пожилого возраста часто становится поводом для выявления опухолей задней черепной ямки или краниоспинальной области. В некоторых случаях правильно поставить диагноз помогают имеющиеся у пациентов внешние проявления: низкая линия оволосения, укороченная шея и т.д., а также наличие на рентгене, КТ и МРТ краниоспинальных признаков костных изменений.
Сегодня "золотым стандартом" диагностики нарушения является МРТ мозга и шейно-грудного отдела. Возможно проведение УЗИ диагностики внутриутробно. К возможным ЭХО-признакам нарушения относятся внутренняя водянка, лимоноподобная форма головы и мозжечок в виде банана. В то же время некоторые специалисты не считают такие проявления специфичными.
Для уточнения диагноза используют различные плоскости сканирования, благодаря чему можно обнаружить несколько информативных симптомов у плода, связанных с болезнью. Получить изображение во время беременности достаточно просто. В связи с этим УЗИ остается одним из основных методов сканирования для исключения патологии у плода во втором и третьем триместрах.
Лечение
При отсутствии симптомов рекомендуется регулярное наблюдение с использованием ультразвукового и рентгенологического исследования. Если единственным признаком аномалии являются незначительные боли, пациентам назначается консервативное лечение. Оно включает различные варианты с применением противовоспалительных препаратов и миорелаксантов. Наиболее распространенными противовоспалительными препаратами являются Ибупрофен и Диклофенак.
Важно не самостоятельно принимать обезболивающие препараты, так как они имеют определенные противопоказания (например, язвенная болезнь). При наличии каких-либо противопоказаний врач подберет альтернативный метод лечения. В некоторых случаях может быть назначена дегидратационная терапия. Если в течение двух-трех месяцев такое лечение не дает результатов, может потребоваться операция (расширение отверстия в затылочной области, удаление дужки позвонка и т.д.). В таких случаях необходимо индивидуальное решение, чтобы избежать ненужного вмешательства или задержки с операцией.
У некоторых пациентов хирургическая ревизия может быть необходима для окончательного постановления диагноза. Цель операции – устранение сдавливания нервных структур и нормализация циркуляции жидкости в спинном мозге. Такое лечение приводит к значительному улучшению состояния у некоторых пациентов. Расширение черепной ямки способствует исчезновению головной боли, восстановлению осязаемости и подвижности.
Благоприятным прогностическим признаком является расположение мозжечка выше первого позвонка и наличие только симптомов, связанных с мозжечком. В течение трех лет после операции могут возникать рецидивы. В таких случаях решение о присвоении инвалидности принимается медико-социальной комиссией.
Частые вопросы
Что такое мальформация Арнольда-Киари 1 типа?
Мальформация Арнольда-Киари 1 типа, также известная как синдром Арнольда-Киари, является редким врожденным нарушением развития задней черепной ямки и спинного мозга. В этом заболевании мозговые структуры, такие как мозжечок, спускаются ниже своего нормального положения и выступают в шейку позвоночника.
Какие симптомы сопровождают мальформацию Арнольда-Киари 1 типа?
Симптомы мальформации Арнольда-Киари 1 типа могут включать головные боли, шум в ушах, проблемы с координацией и равновесием, затруднения с глотанием и речью, проблемы с дыханием, онемение и слабость в руках и ногах, а также изменения в функции мочевого пузыря.
Как диагностируется мальформация Арнольда-Киари 1 типа?
Для диагностики мальформации Арнольда-Киари 1 типа используются различные методы, включая магнитно-резонансную томографию (МРТ), компьютерную томографию (КТ) и рентгенографию. Эти методы позволяют врачам визуализировать аномалии в структуре головного и спинного мозга.
Каково лечение мальформации Арнольда-Киари 1 типа?
Лечение мальформации Арнольда-Киари 1 типа зависит от симптомов и степени тяжести заболевания. В некоторых случаях может потребоваться хирургическое вмешательство для улучшения дренажа спинномозговой жидкости и улучшения симптомов. Дополнительное лечение может включать применение лекарств для управления болевыми симптомами и физиотерапию для улучшения координации и силы мышц.
Полезные советы
СОВЕТ №1
Если у вас была поставлена диагноз мальформации Арнольда-Киари 1 типа, важно обратиться к специалисту – нейрохирургу или неврологу, чтобы получить подробную консультацию и рекомендации по дальнейшему лечению.
СОВЕТ №2
При мальформации Арнольда-Киари 1 типа рекомендуется избегать физической нагрузки, которая может усилить симптомы и вызвать ухудшение состояния. Важно следить за своим здоровьем, регулярно проходить обследования и придерживаться рекомендаций врача.